
Synthesis and Characterization of Coordination Compounds1
Authors: D. Afzal, R. G. Baughman, H. D. Ervin, A. E. Moody, H. D. Wohlers, and J. M. McCormick*
Last Update: March 15, 2013
Introduction
Coordination compounds (also known as complex ions or simply complexes) are formed by the reaction of a Lewis acid (an electron pair acceptor, usually a transition metal) with a Lewis base (an electron pair donor), which is known as a ligand. What is unique about coordination compounds is that they are formed from chemical species that have an independent existence and that this association is often readily reversible (i. e., there is an equilibrium between the solvated metal ion and the ligand). For example, NiCl2 reacts with NH3 in aqueous solution to form the compound Ni(NH3)6Cl2 which contains the complex ion [Ni(NH3)6]2+. This process is easily reversed (by the addition of H+) to give back the starting materials. This type of behavior was thought to be very peculiar by chemists in the 1800’s. They were familiar with compounds like CO2, which although it could be made from C and O2, does not act like it is some loose association of C and O2. It wasn’t until the ground-breaking work of Werner (for which he won the Nobel Prize in chemistry) in the late 19th and early 20th centuries that chemists began to understand these compounds. Werner’s work was greatly expanded on in the 20th century especially after it was discovered that coordination chemistry was relevant to the understanding the role of metal ions in biological systems.
You will be preparing a complex of Co3+ with ethylenediamine, NH2CH2CH2NH2(abbreviated: en), and a complex of Fe3+ with the oxalate ion, C2O42- (abbreviated: ox2-). Ethylenediamine and oxalate are examples of bidentate ligands, which means that they have two different atoms that can donate electron pairs to a metal ion. Ethylenediamine does this through lone pairs on its nitrogen atoms, while oxalate donates electron pairs from two of its four oxygen atoms. In these complexes the metal ion is directly bonded to six other atoms in what is called an octahedral geometry (if we connected the six atoms, the resulting solid would be an octahedron, and hence the name of this geometry). Although it may not be obvious, there may be many ways in which the same six atoms can be arranged around a central atom in an octahedral geometry, and each of these different arrangements may give rise to compounds with the same chemical formula, but have different arrangements of their atoms (isomers). There are many types of isomers: compounds in which the actual connections between atoms (bonds) are different are called constitutional isomers, while compounds where the bonds are the same, but the atoms are arranged differently are called geometric isomers. Geometric isomers are further classified as enantiomers (two compounds that are mirror images of each other) and diastereomers (geometric isomers that are not mirror images).
Because en is a bidentate ligand, the dichlorobis(ethylenediamine)cobalt(III) complex, [Co(en)2Cl2]+, that you will prepare exists as three geometric isomers; one pair of enantiomers and their diastereomer. The isomer where the chlorides are situated on either side of the Co3+ (180° from each other) is called the trans isomer (Fig. 1), while the isomer where the chlorides are next to each other in the octahedron (90° from each other) is the cis isomer. In addition, there are two different ways in which we can put two Cl atoms cis to one another, and these are enantiomers (Fig. 2). The tris(oxalato)ferrate(III) ion, [Fe(ox)3]3-, exists as two enantiomers (which have no diastereomer). In one the three oxalates form a right-handed propeller, and in the other they form a left-handed propeller (both are shown in Fig. 3).
![]() Figure 1. Structure of trans-[Co(en)2Cl2]+ redrawn from the Cambridge Crystal Structure Database entry CENCOS using the Mercury molecular visualization software package.
Figure 1. Structure of trans-[Co(en)2Cl2]+ redrawn from the Cambridge Crystal Structure Database entry CENCOS using the Mercury molecular visualization software package.
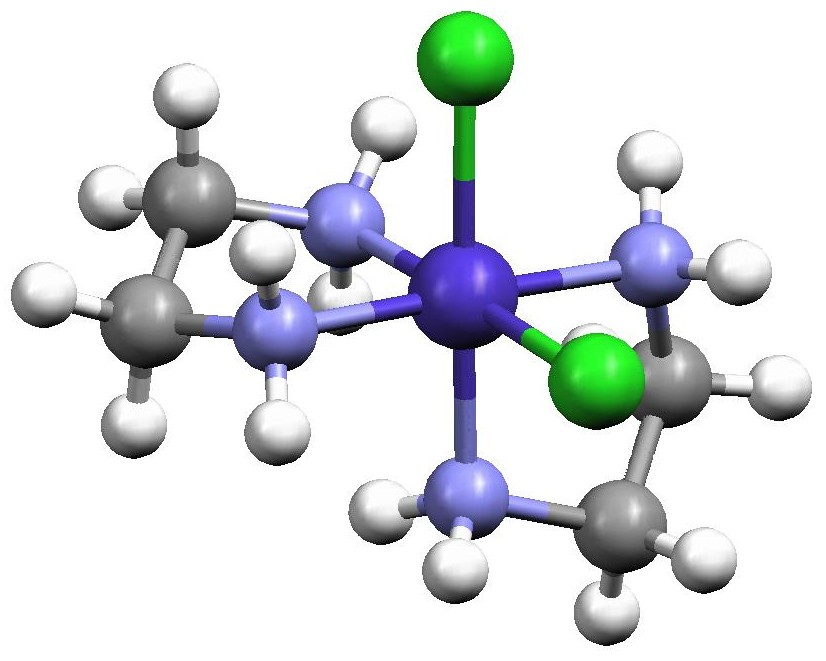
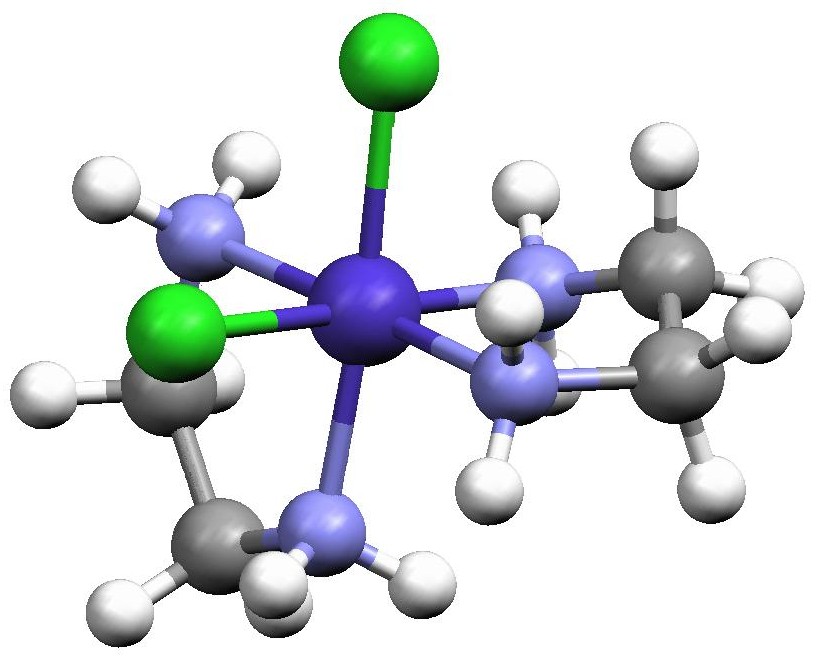 Figure 2. Structures of the two cis-[Co(en)2Cl2]+ enantiomers redrawn from the Cambridge Crystal Structure Database entries CENCOC and CLECOC using the Mercury molecular visualization software package. The isomer on the left is designated as Λ (lambda) while the isomer on the right is designated as Δ (delta) and this is determined by the orientation of the two en ligands. When one en is placed horizontally at the back of the octahedron defined by the six atoms surrounding the Co, the other en cuts across the face of the octahedron either with a positive slope (Λ enantiomer) or a negative slope (Δ enantiomer).
Figure 2. Structures of the two cis-[Co(en)2Cl2]+ enantiomers redrawn from the Cambridge Crystal Structure Database entries CENCOC and CLECOC using the Mercury molecular visualization software package. The isomer on the left is designated as Λ (lambda) while the isomer on the right is designated as Δ (delta) and this is determined by the orientation of the two en ligands. When one en is placed horizontally at the back of the octahedron defined by the six atoms surrounding the Co, the other en cuts across the face of the octahedron either with a positive slope (Λ enantiomer) or a negative slope (Δ enantiomer).
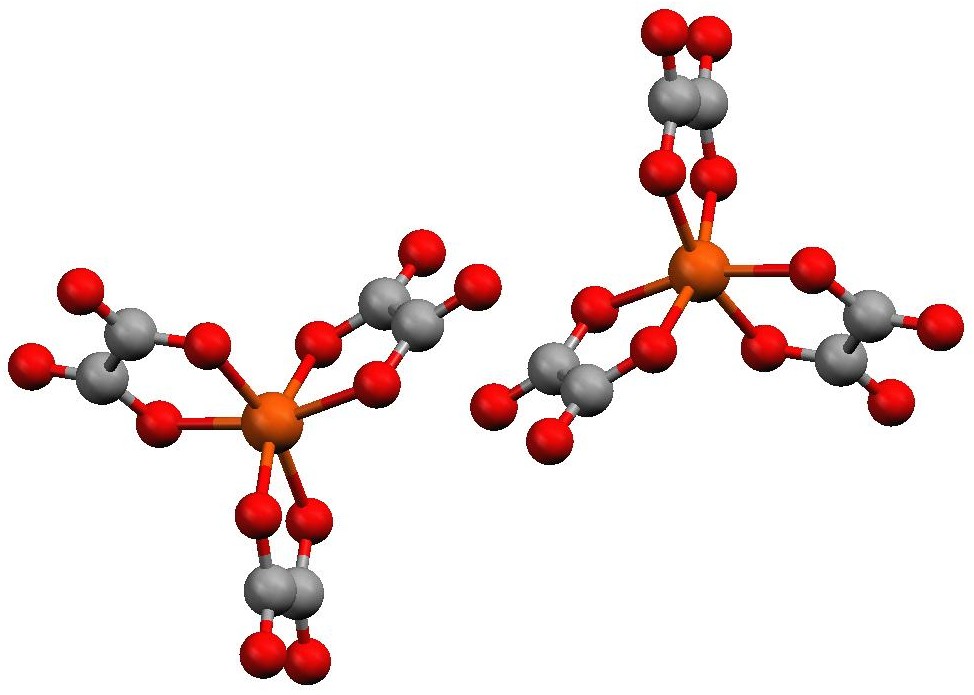 Figure 3. Structures of the [Fe(ox)3]3- enantiomers redrawn using the Mercury molecular visualization software package from data collected by Professor Russell Baughman at Truman State University on crystals prepared by Truman students. The Λ enantiomer is the one on the left and the Δ enantiomer is the one on the right. Unlike the cobalt complex, where both enantiomers can be separated and isolated by crystallization, the enantiomers of the iron complex cannot be separated. This is because there is a rapid pathway available in solution to interconvert the enantiomers.
Figure 3. Structures of the [Fe(ox)3]3- enantiomers redrawn using the Mercury molecular visualization software package from data collected by Professor Russell Baughman at Truman State University on crystals prepared by Truman students. The Λ enantiomer is the one on the left and the Δ enantiomer is the one on the right. Unlike the cobalt complex, where both enantiomers can be separated and isolated by crystallization, the enantiomers of the iron complex cannot be separated. This is because there is a rapid pathway available in solution to interconvert the enantiomers.
Another interesting property of the transition metals is that their reactivity depends on several factors including the metal ion’s charge, the number and type of donor atoms, and the number of d electrons present on the metal ion. It is the later property which we will exploit in this exercise both in the synthesis and then in the reactions of the compounds. The Co2+ ion has a 3d7 electronic configuration and Fe3+ has a 3d5 electronic configuration. Metal ions with these electronic configurations rapidly exchange their ligands and are referred to as labile. The 3d6 Co3+ is an example of an inert ion, that is, an ion that does not rapidly exchange its ligands.
In this experiment, you will synthesize trans-[Co(en)2Cl2]+ from CoCl2·6H2O according to the overall balanced chemical equation given in Scheme 1. In the mechanism by which this compound is formed Co2+ is initially chelated by one en and then a second en, with each step being described by an equilibrium constant. Once two, or more, en have become bound to the Co2+ H2O2 (or, in some cases, O2 from the air) is able to oxidize the metal center to Co3+. Since further substitution of a Co3+ complex is slow, the reaction will essential be over at that point. Thus by careful manipulation of the amount of en present and when the oxidant is introduced, we can force the formation of a Co3+ complex with either two or three en bound to the metal. Because the Co3+ complexes are inert we could subsequently convert trans-[Co(en)2Cl2]+ to cis-[Co(en)2Cl2]+ and then separate (resolve) its enantiomers.
CoCl2·6H2O + 2 en + xs H2O2 + xs HCl → trans-[Co(en)2Cl2]Cl·HCl + other products
Scheme 1. Synthetic route to trans-[Co(en)2Cl2]Cl·HCl starting from CoCl2·6H2O.
Both the cis– and trans-[Co(en)2Cl2]+ complex ions undergo hydrolysis in acidic aqueous solution, as shown in Scheme 2. This reaction can proceed through one of two mechanisms (both are shown in Scheme 3): a dissociative mechanism, where the Cl–leaves the complex before the H2O enters, or an associative mechanism, where the H2O enters the complex before the Cl– leaves.
[Co(en)2Cl2]+ + H2O → [Co(en)2(H2O)Cl]2+ + Cl–
Scheme 2. The hydrolysis reaction of either cis– or trans-[Co(en)2Cl2]+ under acidic conditions. Note that, in general, starting with the trans isomer yields the correspondingtrans isomer and starting with the cis isomer gives the corresponding cis isomer of the product.
| Associative Mechanism |
| [Co(en)2Cl2]+ + H2O → [Co(en)2(H2O)Cl2]+ |
| [Co(en)2(H2O)Cl2]+ → [Co(en)2(H2O)Cl]2+ + Cl- |
| Dissociative Mechanism |
| [Co(en)2Cl2]+ → [Co(en)2Cl]2+ + Cl- |
| [Co(en)2Cl]2+ + H2O → [Co(en)2(H2O)Cl]2+ |
Scheme 3. Possible mechanisms for the acidic hydrolysis of cis– or trans-[Co(en)2Cl2]+.
The problem is further complicated by the interconversion of the cis– and trans– isomers, as shown in Scheme 4. Luckily, this reaction is known to be much slower than the hydrolysis reaction to be studied, and as long as the reaction temperature is not taken much above 70°C for an extended period of time the cis–trans isomerization reaction need not be considered.
trans-[Co(en)2Cl2]+ → cis-[Co(en)2Cl2]+
Scheme 4. Reaction converting trans-[Co(en)2Cl2]+ to cis-[Co(en)2Cl2]+.
In the second week of this exercise, you will determine the activation energy of the first-order hydrolysis of trans-[Co(en)2Cl2]+ under acidic conditions. You will be using a quenching method to study the reaction kinetics (compare to the direct measurement method that was used in the CHEM 130 kinetics exercise, Kinetics of Crystal Violet Bleaching). With a quenching method a small portion of the reaction mixture is withdrawn at specific times and the reaction is then stopped by either adding reagent that stops the reaction (usually by rapidly reacting with one of the reactants) or by rapidly cooling or freezing the sample. For the reaction that we are studying, we will use the latter method to quench the reaction. Each lab group will be assigned different temperatures at which to study the reaction, and from the pooled data you prepare an Arrhenius plot and determine the reaction’s activation energy.
The overall balanced chemical reaction for synthesis of the [Fe(ox)3]3- is shown in Scheme 5. It is the sum of three steps, each corresponding to the stepwise reversible binding of ox2- to Fe3+. The metal remains in the labile 3+ oxidation state throughout the reaction, and so by adding a sufficient amount of ox2-, we can force the reaction essentially completely to [Fe(ox)3]3-. Because the complex is labile, we cannot resolve the [Fe(ox)3]3- enantiomers, but we can use the complex’s lability to confirm the 3:1 ox:Fe3+ stoichiometry.
FeCl3·6H2O + 3 K2C2O4·H2O ® K3[Fe(ox)3]·3H2O + other products
Scheme 5. Synthesis of K3[Fe(ox)3]·3H2O.
In the final week of this exercise, you will confirm the 3:1 oxalate:Fe stoichiometry in your complex in much the same way as Werner did. First you will acidify a solution of [Fe(ox)3]3- which will force the sequential equilibria that formed the complex back to Fe3+(aq) and H2C2O4. The amount of Fe and oxalate present will then be determined by redox titration against a standard KMnO4 solution.
Experimental![]()
For this exercise it is critical that you read each week’s procedure carefully and that you and your lab partner plan what you will do before coming to the laboratory. Each week of this three-week exercise presents different challenges, and unprepared students will find it difficult to finish in the allotted time.
Each pair of students will need to check out the following items from the stockroom: magnetic stir bar, beaker tongs, Büchner funnel and filter flask.
All work of the first week’s work must be carried out in the hood using the two stirring hotplates that will be in each hood. In the cobalt complex synthesis you will use one hotplate ONLY for heating, and the other ONLY for stirring. The stirring hotplate that you used for stirring will then be used in the synthesis of the iron-oxalate compound (this will allow you to start the synthesis of the iron compound while finishing the cobalt compound’s synthesis). There is enough room on each plate for two beakers or two evaporating dishes; work with your hood-mates to assure that everything is done efficiently and safely.
Synthesis of trans-Dichlorobis(ethylenediamine)cobalt(III) Chloride Hydrochloride2,3
Fill a 600-mL beaker approximately half full with water, and place it on a hot plate in the hood. Place your wire gauze on top of the beaker and heat to boiling.
Weigh out approximately 1 g of cobalt(II) chloride hexahydrate, CoCl2·6H2O, measured to the nearest milligram. Place the cobalt salt in your evaporating dish and add 2.5 ml of distilled water. Gently swirl the dish until the cobalt has dissolved.
Once the cobalt chloride has completely dissolved in the water, add 4 mL of 10% ethylenediamine. Place your magnetic stir bar in the solution and set the evaporating dish on a stirring hotplate (NOT the hotplate with your water bath). Set the stir control knob so that the stir bar spins but does not splatter solution on the side of the evaporating dish (a setting of “4” on most stir plates is a good starting point). Stir for 10 min.
After 10 minutes, slowly and carefully add dropwise 1.6 mL of 10% H2O2 to the solution while it is still stirring. CAUTION! 10% H2O2 causes severe skin burns. Gloves are recommended and you should wash your hands after using this solution. Once the H2O2addition is complete, reduce the stir speed (to “3” on most of the laboratory stir plate) and stir for an additional 15 min.
Next, CAREFULLY add 3 mL of concentrated HCl. CAUTION! HCl is VERY corrosive and will cause severe burns. Immediately wash off any HCl that comes in contact with skin with copious amounts of water. Turn off the stir plate and remove the magnetic stir bar using the magnetic stir bar retriever, as your instructor will demonstrate.
Place your evaporating dish on top of the wire gauze on the beaker of boiling water. Reduce the solution’s volume until there is a thick layer of dark green crystals and very little liquid left. This will take between 30 and 45 min. Be sure to monitor the level of boiling water in your water bath, you will need to gradually add water to the water bath during this time to keep it from boiling dry. This is a good point to start the synthesis of the iron complex.
While you are waiting for the volume to be reduced, set up an ice bath. Obtain ~5 mL of methanol and put it in a small beaker or test tube in your ice bath. You will use the ice cold methanol to rinse your crystals in the final step of this synthesis.
Carefully remove the evaporating dish with the beaker tongs and set it on the bench top in the hood to cool. While your solution is cooling, set up the vacuum filtration apparatus (see the CHEM 130 Alum experiment).
Place a piece of filter paper in the Büchner funnel to collect your crystals. Carefully wet the filter paper with a couple drops of methanol BEFORE you add your crystals.
With the aspirator on, transfer the contents of your evaporating dish into the Büchner funnel using a spatula. Add about 2 mL of ice-cold methanol to your evaporating dish, swirl and pour this into the Büchner funnel. This will help remove all of your contents from the evaporating dish. Carefully pour ~5 ml of ice cold methanol over your crystals and filter. Repeat if necessary.
Carefully transfer your crystals to a watch glass, as your instructor will demonstrate. It is not necessary to remove the filter paper at this time. Cover the watch glass with a paper towel and place it in your drawer to dry until next week.
Synthesis of Potassium Tris(oxalato)ferrate(III) trihydrate4,5
In your clean, dry 8 inch test tube dissolve 1.6 g FeCl3·6H2O (measured precisely) in 4 mL of distilled water. It may be convenient to gently heat the solution in a hot water bath on a hot plate to speed the dissolution of larger chunks of the ferric chloride.
Precisely and accurately weight out between 6.0 and 6.5 g K2C2O4·H2O and place it in a clean, dry 50-mL beaker. Add 10 mL of distilled water and gently heat on a hot plate with stirring until all of the potassium oxalate has dissolved. It is not necessary to boil the solution.
Once the oxalate salt has dissolved, quickly and carefully pour the hot oxalate-containing solution into the test tube containing the iron solution. Swirl to mix. Cover and allow the reaction mixture to cool slowly to room temperature. Once the test tube is cool to the touch transfer it to an ice bath and continue cooling.
Green crystals of the product may form during the initial cooling to room temperature, but they might not form until the ice cooling step. If crystals do not form after 30 min of cooling, try gently scratching the bottom of the test tube with a stirring rod or add a seed crystal (a crystal of the product obtained from another preparation), which your instructor will provide.
Decant the solution above the crystals and discard. Recrystallize the crude product by adding approximately 5 to 8 mL of hot distilled water to the crystals in the test tube and heating gently to affect total dissolution. Decant the green solution containing the product into a clean, dry beaker and discard any residue that remains in the test tube.
Cover the beaker with a watch glass and set it aside to cool slowly. When the beaker is cool to the touch, transfer it to an ice bath and continue to cool. Green crystals should form within about 20 min. If they do not, consult your instructor for assistance.
Collect the crystals by vacuum filtration. Wash the crystals twice with 2 mL of ice-cold distilled water and then with two 3-mL portions of acetone. CAUTION! Acetone is flammable, so do all these manipulations in the hood. Dry the product on the filter and then transfer it to a watch glass. Cover the watch glass with a paper towel and place it in your drawer to dry.
Before beginning any work this week obtain the mass of the dry trans-dichlorobis(ethylenediamine)cobalt(III) chloride hydrochloride and the dry potassium tris(oxalato)ferrate(III) trihydrate.
Determination of the Activation Energy for Hydrolysis of trans-Dichlorobis(ethylenediamine)cobalt(III) Chloride6,7
You will be using the Ocean Optics spectrometers to monitor the progress of the reaction. Please review the operation of these spectrometers and the basics of kinetics in the CHEM 130 lab exercise Kinetics of Crystal Violet Bleaching (Kinetics of Crystal Violet Bleaching) before coming to lab.The Ocean Optics spectrometer will be already turned on and warmed up by the time you arrive in the laboratory. Do NOT unplug the spectrometer from the computer, shut down either the spectrometer or the computer, or make any changes to the experimental set-up. Doing so will require a time-consuming complete re-start of the system.
At some point before beginning your kinetics runs you will need to calibrate the spectrometer using a cuvette filled with distilled water. Important! you must use the same cuvette for both the blank and for the actual measurements. Before inserting the cuvette into the spectrometer be sure to thoroughly wipe the cuvette’s windows with a KimWipe. Any bubbles adhering to the cuvette’s windows may be dislodged by gentlytapping the cuvette with your finger. Do NOT tap the cuvette on the table! Be sure that the cuvette’s clear windows are aligned perpendicular to the spectrometer’s long sidewhen you insert it into the spectrometer. Also be sure that the cuvette is inserted into the spectrometer as reproducibly as possible each time. Many problems that students experience with this exercise can be traced to the cuvette not being cleaned and not being inserted properly into the instrument.
Check out a thermometer and obtain several plastic centrifuge tubes from the stockroom.
Be sure that your large test tube and all of your small test tubes are clean and dry. You will be running the reaction in the large test tube and using the small test tubes to store the aliquots removed from the reaction mixture. Place 3 ml of distilled water in each of your small test tubes and mark the water level; discard the water and dry the test tubes.
Each bench will be assigned two temperatures at which to run the reaction (the pairings are usually 40/65 °C, 45/60 °C and 50/55 °C with two benches in each lab section running the same temperature pairing, but your instructor will inform you of your actual assignments in the laboratory). Water baths set for each of these temperatures will be in the hoods. They will be labeled and your instructor will point out which bath is in which hood. Do NOT change the settings on any bath! Use the thermometer to find the actual bath temperature to the nearest 0.1 °C, but do NOT leave the thermometer in the water bath between measurements and be careful where you set it between measurements. Note: it is not critical that you are exactly at your assigned temperature, as long as the temperature reading is stable and you record and use the actual temperature at which the data were collected.
The actual logistics of how you and your bench mates will accomplish your assigned measurements will depend on the number of groups in the lab and the number of available spectrometers. Your instructor will give you guidance, but it is essential that you and your bench mates develop a plan of attack before coming to the laboratory.
Place 30 ml of 0.01 M HNO3 in your large test tube and place it in the water bath. Place 1 ml of cold distilled water in a small centrifuge tube (the tubes can contain up to about 3 ml and the volumes are marked on the side). Keep the centrifuge tube cold in an ice bath until ready for use.
To perform a kinetics run, precisely weigh out approximately 0.2 g of trans-[Co(en)2Cl2]Cl·HCl. Place this in the small centrifuge tube containing the cold water, close the tube and shake vigorously to dissolve the solid. Pour the entire contents of the centrifuge tube into your large test tube and mix thoroughly. Do this as quickly as possible; it is best if this can be accomplished without removing the large test tube from the water bath. Start timing from when the solution is completely mixed.
Remove approximately 3 ml of the reaction mixture immediately upon mixing. Place it in one of your small test tubes and place the tube in the ice bath. Obtain the absorbance spectrum of this sample once it has cooled back to about room temperature and record the absorbance at 505 nm and at 800 nm (this is your data point at t = 0). Don’t forget tothoroughly wipe the cuvette’s windows with a KimWipe before you place it in the spectrometer. Any bubbles adhering to the cuvette’s windows may be dislodged bygently tapping the cuvette with your finger. Do NOT tap the cuvette on the table! If the absorbance measurements at 800 nm is not approximately zero, re-wipe the cuvette’s windows and try the measurement again.
You will repeat this process until you have removed a total of nine samples. The timing between samples will vary with temperature. At 40 °C you must follow the reaction for at least 40 minutes, but at 65 °C you will only need to follow the reaction for 5 minutes. Work out the timing of the sample withdrawals before you start a run.
Once you have removed nine samples, place the test tube containing the remaining 3 ml of the reaction mixture in the 70-°C water bath at for no more than five minutes. Remove the test tube from the bath and place it in the ice bath. Obtain the reaction mixture’s absorbance spectrum, and record the absorbance at 505 nm and 800 nm, once it has cooled to near room temperature; this is your t = ∞ data point.
Prepare a fresh sample and obtain data at the other assigned temperature. Repeat the measurements at the two temperatures as time permits.
All cobalt-containing waste is to be placed in the proper container. Any unused cobalt complex will be saved for later use. Please put it in the container provided, as directed by your instructor.
Week 3
You will need to write balanced chemical equations for the two different redox reactions that you will use in this analysis. Do this before coming to the lab. To assist you in this note that: 1) the reduction of MnO4– in acidic solution gives Mn2+ as a product, 2) the oxidation of H2C2O4 or C2O42- in acidic solution gives CO2 as a product, 3) reduction of Fe3+ gives Fe2+ (and so the oxidation of Fe2+ gives Fe3+), and 4) oxidation of Zn gives Zn2+. There are other reactants and products (e. g., H+ and H2O) that you will need to add to balance the redox reactions. Before coming to lab determine how much K2C2O4·H2O is needed so that, when it is titrated with a 0.01 M MnO4– solution, the equivalence point is reached at 20.00 mL of added titrant. Set up this and any other calculations that you will need to determine the % C2O42- and % Fe by mass in the iron complex from your titration data. Be aware that students who come to lab ill-prepared will most likely not finish the exercise.
Standardization of 0.01 M Potassium Permanganate Solution
Obtain approximately 75 mL of un-standardized 0.01 M KMnO4 solution. CAUTION!Potassium permanganate is a strong oxidizing agent! Immediately wash any spilled on your skin with copious amounts of water.
Wash your buret several times, as your instructor will demonstrate, with no more than 2 mL of the permanganate solution. Each time allow the solution to drain through the buret’s tip into a waste beaker. After the final wash fill the buret to near the 0.00-mL mark such that the upper part of the meniscus is at or below the 0.00-mL mark. Make sure that there are no bubbles in the buret tip. Run some of the solution through to clear any bubbles (gentle taping might help dislodge any recalcitrant bubbles). Refill the buret, if necessary. Record the initial volume of solution in the buret. Because the MnO4– ion is so strongly colored, make all your volume readings from the top of the meniscus.
Accurately weigh out the amount of K2C2O4·H2O that you calculated you will need to standardize the MnO4– solution (helpful hint: it should be around 0.1 g). Place the oxalate in an Erlenmeyer flask and add about 30 mL of distilled water and 5 mL of 6 M H2SO4. Swirl the flask gently to dissolve the solid.
Place the flask on a stirring hotplate in the hood and carefully heat the solution to approximately 60 °C. Do not boil! Remove the flask from the hotplate (a folded paper towel makes an excellent hot pad). This titration must be performed fairly quickly so that the solution does not cool too much. Be sure that you swirl the flask as you add the permanganate solution and wash down any solution that splashes on the side of the flask with a small stream of distilled water. If the solution cools too much, reheat. Titrate the sample with the permanganate solution until the slightest purple color persists for at least 30 sec. Record the volume of permanganate used and from it determine the [MnO4–]. Repeat until three determinations agree with each other within 2%.
Determination of Oxalate in Potassium Tris(oxalato)ferrate(III) Trihydrate
Accurately weigh out about 0.10 g of your K3[Fe(ox)3]·3H2O, place in an Erlenmeyer flask, add 30 mL distilled water and 5 mL of M H2SO4. Swirl to dissolve the solid and then heat to 60 °C, again taking care not to boil the solution. Titrate as described above to the first permanent purple color. Record the volume of titrant dispensed and from it calculate the % C2O42- by mass in the sample.
Do NOT discard the solution at this point; proceed to the iron analysis given below.
Determination of Iron in Potassium Tris(oxalato)ferrate(III) Trihydrate
Take your faintly-purple sample from the oxalate analysis above, and set in on a hotplate in the hood (there may be a small amount of brown precipitate forming at this point, but do not be concerned). Gently heat it until it almost boils and add approximately 100 mg of Zn to the hot solution. Cover the flask with a watch glass and continue heating until the yellow color (from Fe3+) disappears (Fe2+ is colorless in solution). While you are waiting, set up a gravity filtration with fluted filter paper (ask your instructor to demonstrate how to flute filter paper, if you are unsure).
Quickly filter the hot, colorless solution into another Erlenmeyer flask, again using a folded paper towel to handle the hot flask. It is important that this filtration be done quickly to minimize the amount of Fe2+ that is re-oxidized to Fe3+ by O2 in the air. Rinse the funnel with several small (approximately 5 mL total volume) portions of distilled water. Titrate this solution with the permanganate solution in your buret until the first faint trace of a persistent pink color. Record the volume of permanganate used and from it determine the % Fe by mass in your sample
Repeat the two titrations until you run out of sample, or when three of the oxalate titrations agree with each other within 2%.
Results and Analysis
Week 1
After you have determined the mass of your dry trans-[Co(en)2Cl2]Cl·HCl and K3[Fe(ox)3]·3H2O, calculate a percent yield for each compound. In each case assume that the metal salt is the limiting reagent. Note that this is a common practice among coordination chemists because of all the coupled equilibria involved in the synthesis of coordination compounds.
Week 2
Prepare a graph in Excel showing absorbance as a function of wavelength in which you overlaid all of the spectra for one run. The scale on the y-axis should be 0 to 1 (you may need to set the maximum value on the y-axis to 1.1 or 1.2, depending on your actual concentrations) and the x axis scale should be set from 400 to 800 nm. If the data were obtained correctly, the spectra should all have an absorbance of about 0 at 800 nm and there should be two isosbestic points (points where the absorbance does not change as a function of time, indicating that only two species were contributing to the absorbance throughout the reaction).
From the absorbance at 505 nm at each time subtract the absorbance at 800 nm at the same time; this corrects for any baseline drift or other variations that occurred between samples. With the corrected absorbance readings at 505 nm (At) and your corrected absorbance at infinite time (A∞), prepare a first-order integrated rate law graph (i. e., ln(A∞ – At) as a function of time) for each run. We use A∞ – At instead of concentration in these graphs because 1) we are following the formation of the product over time and 2) the reactant has a relatively large absorbance at this wavelength.
From each graph determine the value of the rate constant at that temperature. Report each rate constant (along with its uncertainty at 95% confidence) and the temperature at which it was obtained to the instructor and the rest of the class. From the class data (rate constant as a function of temperature), prepare an Arrhenius plot, determine the activation energy and its confidence interval at the 95% confidence limit.
Does the fact that the hydrolysis reaction is first-order in the cobalt complex and your value of the activation energy allow you to conclusively rule out one of the possible mechanisms? You will need to derive the rate law predicted by each mechanism for each of the possible cases: 1) the first step is irreversible and rate determining, 2) the first step rapidly goes to equilibrium and the second step is slow, 3) the first step is reversible, but does not come to equilibrium and the second step is slow. What additional experiments could you do to help distinguish between the possible mechanisms?
Week 3
Calculate the average percent oxalate by mass in your K3[Fe(ox)3]·3H2O and share this with the class. From the class data calculate the average percent oxalate by mass in K3[Fe(ox)3]·3H2O, the standard deviation and the confidence interval at the 95% confidence limit. Be sure to perform a Q-test on the class data first to exclude any suspect point. Calculate the true percent oxalate by mass and determine a percent error for both your data and the class data.
Calculate the average percent iron by mass in your K3[Fe(ox)3]·3H2O. Share this with the class, and calculate the average percent iron by mass in K3[Fe(ox)3]·3H2O, the standard deviation and the confidence interval at the 95% confidence limit from the class data. Perform a Q-test on the class data first to exclude any suspect point. Calculate the true percent iron by mass and determine a percent error for both your data and the class data.
Conclusions
This exercise contains elements of both a synthesis exercise and a measurement exercise. Your instructor may have you treat all three weeks as one exercise for the purposes of writing your Discussion of Conclusions, or he/she might have you treat each week separately. If you are unsure, ask! In addition to the points covered in the synthesis and measurement outlines, be sure to address the questions raised in the Results and Analysis section above.
Summary of Results
In your Summary of Results you should have tables that summarize the results for each week: Week 1, the percent yield of trans-[Co(en)2Cl2]Cl·HCl and K3[Fe(ox)3]·3H2O; Week 2, the average rate constants at each temperature that you determined and the activation energy that was determined from the class data (along with a 95% confidence interval for each, if possible); and Week 3, the statistical (average, standard deviation and confidence interval) results for the K3[Fe(ox)3]·3H2O analyses.
References
- Click here to download this file in PDF format. Note that hyperlinks are not active in the pdf version.
- Szafran, Z.; Pike, R. M. and Singh, M. M. Microscale Inorganic Chemistry: a Comprehensive Laboratory Experience; John Wiley and Sons: New York, 1991.
- Bailar, J. C., Jr. Inorg. Synth. 1946, 2, 222-225.
- Beran, J. A. Laboratory Manual for Principles of General Chemistry, 5th Ed.; John Wiley and Sons: New York, 1994.
- Marcus, S.; Sienko, M. J. and Plane, R. A. Experimental General Chemistry; McGraw-Hill: New York, 1988.
- Jolly, W. L. Encounters in Experimental Chemistry, 2nd Ed.; Harcourt, Brace, Jovanovich: New York, 1985, 77-79.
- Herrick, R. S.; Mills, K. V. and Nestor, L. P. J. Chem. Educ. 2008, 85, 1120-1122. Click here to view a PDF version of this article (Truman addresses and J. Chem. Educ. subscribers only).