
Variation of K for the Keto-Enol Tautomerism of ß-Dicarbonyls
Author: J. M. McCormick
Last Update: January 8, 2015
Introduction
The enhanced acidity of the alpha protons, and the even more enhanced acidity of the alpha protons in ß-dicarbonyls, is an important feature of carbonyl chemistry.1 The carbonyl oxygens of the ß-dicarbonyls can act as proton acceptors leading to two distinctly different forms of the dicarbonyl, the keto and the enol, which are in equilibrium with each other, as shown in Scheme 1.1 Although there evidence that, at least for some ß-dicarbonyls, that the enol exists as two rapidly equilibrating forms rather than a pair of equally contributing resonance structures.2,3 Whether this equilibrium favors the keto or the enol form depends on the degree to which the different forms can be stabilized, either internally (i. e., steric hindrance)4 or externally (i. e., solvent-carbonyl interactions).4,5
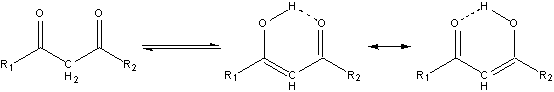
Scheme 1. Keto-enol tautomerism of a generic ß-dicarbonyl.
In this exercise you will investigate the keto-enol tautomerism of several ß-dicarbonyls. You will use NMR spectroscopy to determine the equilibrium constant for the reaction, and then investigate the reasons for any differences in the equilibrium constants via computational methods.
Procedure
Read this entire packet, the procedure in Garland et al.,6 and the modifications suggested by Grushow and Zielinski,7 before developing your own experimental protocol. You must have a clearly defined goal for this experiment, which must be stated in your laboratory notebook’s Statement of Purpose. Groups that do not seem to have a clearly defined goal will not be allowed to start until they articulate one. Groups that seem to be trying every possible combination of solvent and ß-dicarbonyl in the hope they will find something will be made to stop and reevaluate their goals.
We have the following deuterated solvents available: acetone, acetonitrile, chloroform, dimethylsulfoxide, water, and benzene. Note that some of these solvents are very expensive and will only be available after you have demonstrated competence with the less expensive solvents (i. e., CDCl3). At present we have available acetylacetone (2,4-pentanedione), ethyl acetoacetate, methyl acetoacetate, diethyl malonate, t-butyl acetoacetate and ethyl benzoylacetate for analysis. Our NMR, unlike the one described in Shoemaker, does not require TMS or an external lock as long as deuterated solvents are used. In general, your sample needs to have a volume of 0.5-0.7 ml of deuterated solvent (approximately 1 inch in our NMR tubes) and 1-3 drops of the analyte (for 1H NMR). However, you need to determine the molality of the solution to the highest precision you can given the small volumes required.
You will obtain the 1H of each sample in each different solvent and assign all the peaks. Although you may use online and other databases for assistance, you must assign each spectrum yourself. If necessary, obtain the 13C and DEPT-135 spectrum of any sample where the peak assignments are unclear to aid in the assignment.
When you have finished with a sample, dispose of it in the proper waste bottle. Rinse the NMR tubes copiously with acetone (place the acetone in the appropriate waste bottle) and allow them to dry before returning them to the instructor. Be sure that you are finished with a sample before discarding it, because the cost of some of the reagents means that you will have only one shot at some samples.
Click here to proceed to the Truman WebMO9 site which is our portal for Gaussian.10 For the computational component follow the method described by Polik and Schmidt in exercise 32 of the WebMO User’s Guide.9b It is important that you exactly follow their procedure for your molecules (helpful hint: to delete a bond, simply select it and then hit the delete key in the molecular editor). You can obtain thermodynamic information for most levels of theory by performing a vibrational calculation (note that the units will be Hartrees per molecule, not kJ/mole and that the value is for the gas phase). You should also note that the level of theory that these authors suggest (PM3) was chosen because the calculations can be completed quickly; the answers may not be particularly accurate. You may wish to explore whether other theories (or basis sets within PM3) will give a more accurate value of ΔG (and K) for your system. You might try density functional theory (e. g., B3LYP), Hartree-Fock, or one of the theories used in the HCl exercise. Just be aware that there is always a compromise between speed and accuracy in computational chemistry.
Results and Analysis
Data analysis must be performed with the Bruker TopSpin software on the NMR computer.
Show typical spectra in your results section along with a description of how the peaks were assigned (you must demonstrate that you made your own assignment). Determine the equilibrium constant for the keto-enol tautomerization reaction in each solvent by integrating the appropriate peaks in the 1H NMR. Note that in most of the systems under consideration you will be able to calculate this using more than a single pair of peaks, and therefore you can determine an average value of K for each compound along with a confidence interval. Compare your results to previously reported values8 and to the gas phase data.6 Do the computational results match what the expected mechanism? Do the computational results give you any insight into any trends in the equilibrium constants? Is the calculated Ea for the keto-enol tautomerism consistent with what you observed in the NMR? Address whether your data supports or refutes your hypothesis and what other experiments your results suggest that you do next.
1. Solomons, T. W. G. Organic Chemistry, 4th Ed.; Wiley: New York, 1988, p. 783-810.
2. Matsuzawa, H.; Nakagaki, T. and Iwahashi, M. J. Oleo. Sci. 2007, 56, 653-658. Click here to view this article.
3. Perrin, C. L. and Kim, Y.-J. J. Am. Chem. Soc. 1998, 120, 12641-12645. Click here to view this article (Truman addresses and J. Am. Chem. Soc. subscribers only).
4. Folkendt, M. M.; Weiss-Lopez, B. E.; Chauvel, J. P. and True, N. S. J. Phys. Chem. 1985, 89, 3347-3352. Click here to view this article (Truman addresses and J. Phys. Chem. subscribers only).
5. Powling, J. and Bernstein, H. J. J. Am. Chem. Soc. 1951, 73, 4353-4356. Click here to view this article (Truman addresses and J. Am. Chem. Soc. subscribers only).
6. Garland, C. W.; Nibler, J. W. and Shoemaker, D. P. Experiments in Physical Chemistry, 7th Ed.; McGraw-Hill: New York, 2003, p. 453-461.
7. Grushow, A. and Zielinski, T. J. J. Chem. Educ. 2002, 79, 707. Click here to view this article (Truman addresses and J. Chem. Educ. subscribers only).
8. Burdett, J. L. and Rogers, M. T. J. Am. Chem. Soc. 1964, 86, 2105. Click here to view this article (Truman addresses and J. Am. Chem. Soc. subscribers only).
9a. Polik, W. F and Schmidt, J. R. WebMO Pro 6.1.010p; WebMO LLC: Holland, MI, 2006.
b. Polik, W. F. and Schmidt, J. R. WebMO User’s Guide; 2003,http://www.webmo.net/download/WebMO_Users_Guide.pdf.
10. Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; Scuseria, G. E.; Robb, M. A.; Cheeseman, J. R.; Montgomery, Jr., J. A.; Vreven, T.; Kudin, K. N.; Burant, J. C.; Millam, J. M.; Iyengar, S. S.; Tomasi, J.; Barone, V.; Mennucci, B.; Cossi, M.; Scalmani, G.; Rega, N.; Petersson, G. A.; Nakatsuji, H.; Hada, M.; Ehara, M.; Toyota, K.; Fukuda, R.; Hasegawa, J.; Ishida, M.; Nakajima, T.; Honda, Y.; Kitao, O.; Nakai, H.; Klene, M.; Li, X.; Knox, J. E.; Hratchian, H. P.; Cross, J. B.; Adamo, C.; Jaramillo, J.; Gomperts, R.; Stratmann, R. E.; Yazyev, O.; Austin, A. J.; Cammi, R.; Pomelli, C.; Ochterski, J. W.; Ayala, P. Y.; Morokuma, K.; Voth, G. A.; Salvador, P.; Dannenberg, J. J.; Zakrzewski, V. G.; Dapprich, S.; Daniels, A. D.; Strain, M. C.; Farkas, O.; Malick, D. K.; Rabuck, A. D.; Raghavachari, K.; Foresman, J. B.; Ortiz, J. V.; Cui, Q.; Baboul, A. G.; Clifford, S.; Cioslowski, J.; Stefanov, B. B.; Liu, G.; Liashenko, A.; Piskorz, P.; Komaromi, I.; Martin, R. L.; Fox, D. J.; Keith, T.; Al-Laham, M. A.; Peng, C. Y.; Nanayakkara, A.; Challacombe, M.; Gill, P. M. W.; Johnson, B.; Chen, W.; Wong, M. W.; Gonzalez, C. and Pople, J. A. Gaussian 03, Revision C.02; Gaussian, Inc.: Wallingford, CT, 2004.