
Hydrolysis of Oil of Wintergreen1
Authors: D. Afzal, A. E. Moody and J. M. McCormick*
Last Update: January 15, 2013
Introduction
Oil of wintergreen is an essential oil obtained from wintergreen leaves or sweet birch bark. The primary constituent of oil of wintergreen is methyl salicylate (its structure is shown in Fig. 1), which has a fragrant smell and is responsible for the wintergreen flavor in foods and beverages. Because of the high demand in the food industry for methyl salicylate most of it is made synthetically, which is both cheaper and easier than extracting it from the natural sources.

Figure 1. Structure of methyl salicylate.
Methyl salicylate undergoes hydrolysis in the presence of H+ or OH-. An hydrolysis reaction is where something is broken apart by water (hydro– = water, –lysis = splitting). In the experimental procedure that you will follow, the methyl salicylate will be first reacted with a concentrated NaOH solution (the source of OH–) to give compound Y, which we will not isolate (compound Y is an example of a synthetic intermediate). A sulfuric acid (H2SO4) solution will be added as a H+ source to convert compound Y into compound X, which we will collect and characterize. The reaction’s two steps are shown in Scheme 1.
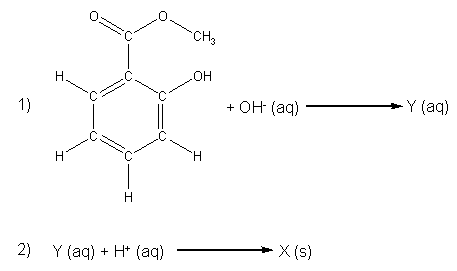
Scheme 1. Unbalanced chemical equations for the hydrolysis of methyl salicylate to give compound X.
When these two steps are added together, we get the overall chemical equation for the hydrolysis of methyl salicylate shown in Scheme 2 (More Info). It is important for you realize that none of the reactions in either scheme are complete! We are missing products, and we do not know the identity of either compound Y or compound X.
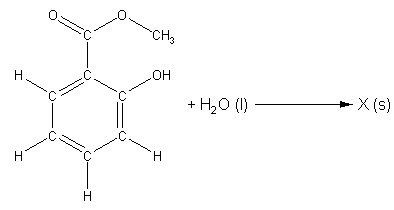
Scheme 2. Overall unbalanced chemical equation for the hydrolysis of methyl salicylate.
You will hydrolyze oil of wintergreen in Week 1 of this exercise and then in Week 2 you will use chemical, physical and spectroscopic means to identify compound X. First you will demonstrate that compound X is an acid, and then you will titrate a known amount ofX with a standardized base to determine its molar mass. You will also compare the melting point of impure and recrystallized X. Pure substances have unique and distinct melting points, while impure substances (mixtures) usually do not have a unique melting point; rather they melt over a range of temperatures. Thus, a melting point determination is a quick and easy way to determine the purity of a substance, if its melting point is neither too high (as is the case with many ionic compounds) nor too low (as for most gases).
In this exercise and in the previous one, we have seen how compounds are characterized by their chemical and physical properties, and until the 1960’s these were the primary ways to characterize new compounds. Starting in the 1960’s new methods, based on the interaction of matter with electromagnetic radiation, revolutionized chemistry. These spectroscopic techniques were faster than the older methods and gave much more information on the substances being analyzed, and so they have almost entirely supplanted the older methods. In this exercise you will be introduced to one of the most widely used and powerful spectroscopic techniques, nuclear magnetic resonance (NMR) spectroscopy.
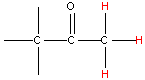
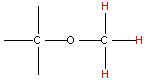
NMR spectroscopy uses the fact that certain nuclei behave like very small magnets, which in a magnetic field can either line up with the field or against it. The alignment of the nuclei can be flipped when they absorb electromagnetic radiation of the correct frequency (in NMR spectroscopy the frequency is expressed as a chemical shift with units of parts per million, ppm). The frequency of radiation that is needed to perform this flip depends on the nucleus and the environment around the nucleus. And so, information on how atoms in molecules and polyatomic ions are arranged and the bonding interactions between them can be determined. Typical chemical shift values for various arrangements of hydrogen and carbon atoms are shown in Table 1. Note that these are typical values and that an atom’s chemical shift might be outside the stated range depending on its environment and what is atoms are bonded to it.
| Type of Hydrogen (in red) | Typical Chemical Shift (ppm) | Type of Carbon (in red) | Typical Chemical Shift (ppm) |
|---|---|---|---|
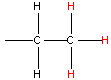 | 0.9 |  | 13 - 16 |
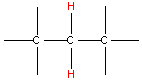 | 1.3 - 1.5 | 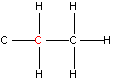 | 16 - 25 |
 | 1.5 - 2.0 |  | 25 - 38 |
 | 2.0 - 2.3 | 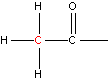 | 20 - 30 |
 | 3.8 | 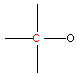 | 50 - 90 |
 | 6 - 8 |  | 125 - 130 |
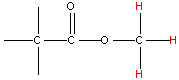 | 3.8 | 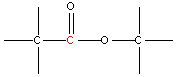 | 170 - 175 |
 | 9 - 11 | 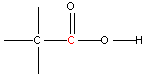 | 177 - 185 |
 | 6 - 12 |  | 205 - 210 |
Table 1. Typical chemical shifts for selected hydrogen atoms in 1H NMR spectra (left) and carbon atoms in 13C NMR spectra (right). In each entry the chemical shift is for the atom shown in red.2
The 1H nucleus (protium) and the 13C nucleus are the most common nuclei studied with NMR spectroscopy. Protium is the most abundant isotope of hydrogen, and so it is likely that in any particular molecule two or more 1H nuclei are near enough to each other to interact. This interaction between 1H nuclei leads to a splitting of the NMR line for each 1H nucleus (see Fig. 2). The splitting is predictable, and can be used to determine the molecule’s structure, but it will not be discussed further in this class.
Because the natural abundance of 13C is low, it is unlikely that two 13C nuclei will be next to each other, so we do not need to worry about the interaction between 13C nuclei. A 13C nucleus can, however, interact with nearby 1H nuclei and this interaction leads to a splitting of the 13C line. However, the 13C NMR experiment is usually set up such that this interaction between the 1H and 13C nuclei is suppressed. The result is that a 13C NMR spectrum consists of a set of lines, each corresponding to a unique carbon in the molecule.
A final piece of information that we can extract from a 1H NMR spectrum is the number of hydrogens present. We do this by integrating the area under a peak, which is proportional to the number of hydrogen atoms that flip at a particular energy. This integration gives the relative number of hydrogens, but not the absolute number, so it is similar to an empirical formula. A 13C NMR spectrum cannot be integrated, because of the way the experiment is performed, but we can determine the number of different carbon atoms present in the molecule by simply counting the number of peaks in the spectrum.
There is one serious problem with NMR spectroscopy. Consider this: if we are trying to find a 1H NMR signal from a solute dissolved in a solvent that contains 1H, how are we ever going to find it in the midst of all the solvent’s 1H? The answer is “not easily.” To avoid this problem NMR spectra are obtained in solvents where 1H have been replaced by 2H (deuterium, often given the atomic symbol “D”). Although 2H has an NMR signal, it is at a different frequency than that of 1H, and doesn’t interfere. Because 100% deuterated solvents are seldom used, there are usually small peaks in the spectrum corresponding to residual un-deuterated solvent that can be ignored in the analysis of the spectrum.
The 1H NMR spectrum of methyl salicylate is shown in Fig. 2. We see six signals; two are single, un-split peaks (called singlets in NMR jargon) and four are multiple peaks (called multiplets). Click on the peaks in Fig. 2 to see more information on them and an enlarged view of the multiplets in the 6 to 8 ppm range. The singlet at 3.927 ppm integrates as 3 hydrogens, and so it must arise from the hydrogens in the (C=O)-O-CH3group of atoms. The peaks in the 6 to 8 ppm range each integrate as 1 hydrogen and their chemical shift is correct for the hydrogens on the ring of carbons (called an aromatic ring). That leaves only the singlet at 10.766 ppm, which integrates as 1 hydrogen, that must be the hydrogen in the -OH group attached to the ring.
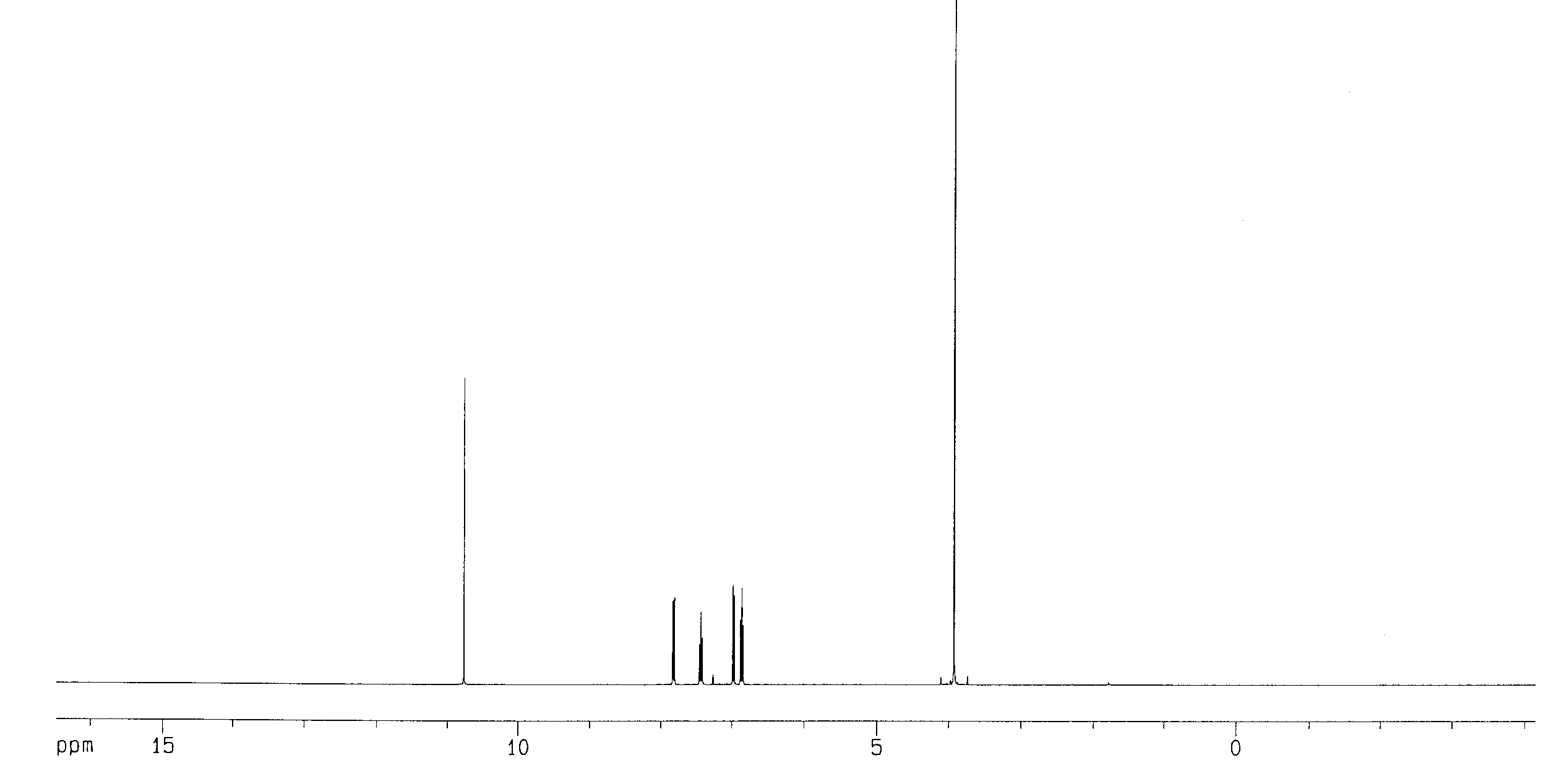
Figure 2. 1H NMR spectrum of methyl salicylate.
Methyl salicylate’s 13C NMR spectrum is shown in Fig. 3. Counting signals we find 8 singlets and 1 multiplet. The multiplet (near 79 ppm) arises from residual non-deuterated chloroform, while the other signals arise from the eight unique carbons in the molecule. The peak at 52.167 ppm is the carbon in the –CH3 group, while the five peaks between 110 and 140 ppm are five of the six ring carbons based on their chemical shifts. The peak at 170.533 ppm is in the range expected for a carbon double bonded to one O and singly bonded to both a C and another O. This leaves the peak at 161.622 ppm as the remaining ring C (note that it is slightly outside the expected range, but we really can’t assign it any other way).
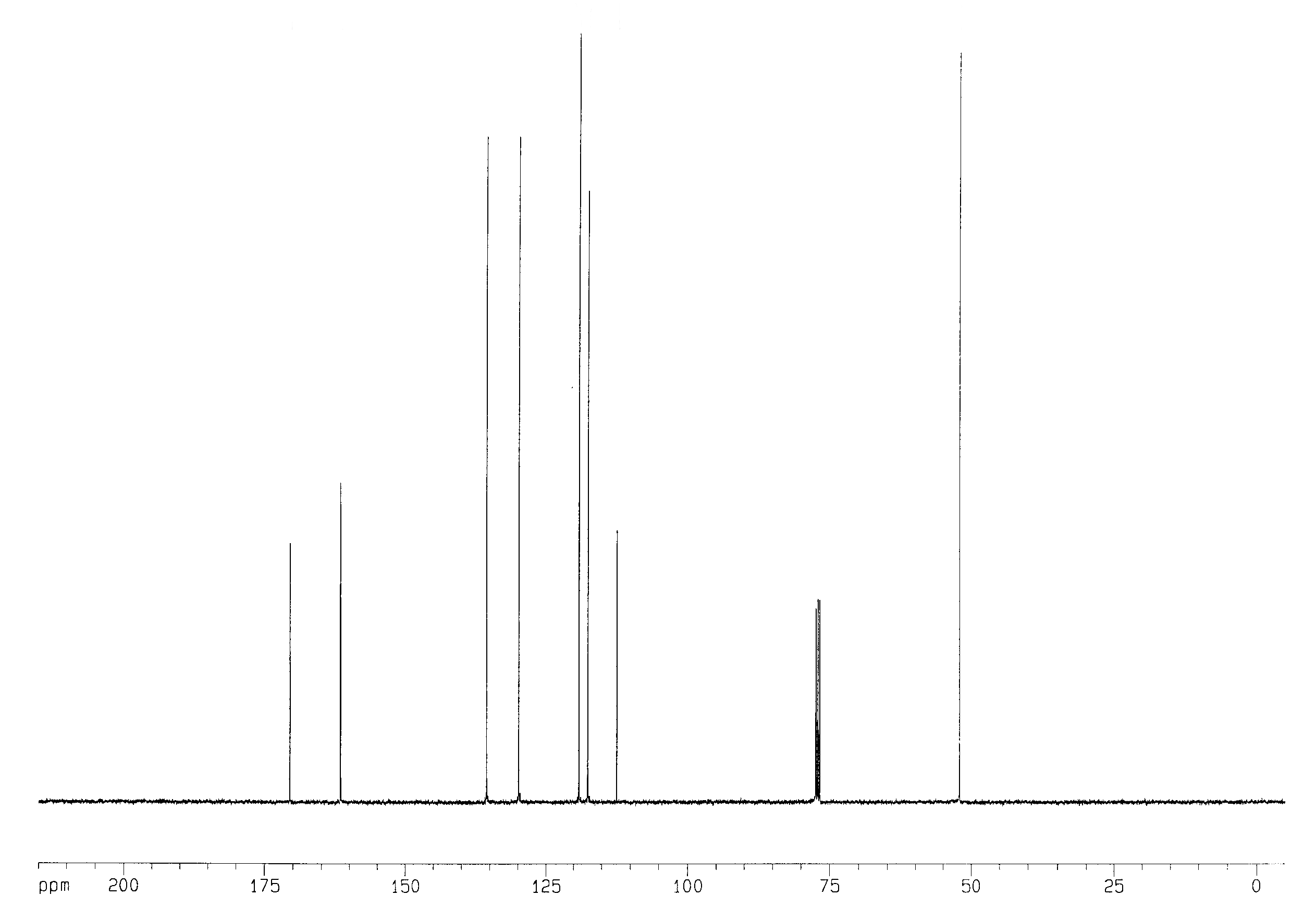
Figure 3. 13C NMR spectrum of methyl salicylate in CDCl3. The multiplet near 77 ppm arises from residual CHCl3.
Click here to download a PDF file containing the NMR spectra of methyl salicylate in a printable format.
In your laboratory notebook, treat both weeks of this exercise as one experiment. That means you will have one Statement of Purpose, one Procedural Outline (this may be a bit long), one Results section, one Calculations section and one Discussion of Results and Conclusions. Don’t worry about preparing a discussion of the first week’s results. Wait until you have all of the results at the end of the second week before you start trying to make sense of everything (it will be a lot easier!).
Experimental 3-5![]()
Place 50 mL of water and a magnetic stir bar (or a few boiling stones) in a 250 mL Erlenmeyer flask. Carefully add 10 g NaOH and swirl to dissolve. CAUTION! Sodium hydroxide is very caustic (causes burns) and the solution will become quite hot (dissolution of NaOH is exothermic). Once the solution has cooled add 5 mL (measured to the nearest 0.1 mL) of oil of wintergreen. Record this volume in your notebook and calculate the mass of oil of wintergreen added from its density (1.18 g/mL).
A white solid may appear when the oil of wintergreen is added to the NaOH solution, but will disappear as the solution is heated. Place a watch glass on top of the Erlenmeyer flask. This will provide a place for water vapors to condense and fall back into the flask, maintaining the level of liquid in the reaction (this is called refluxing). Covering the flask also prevents splashing of the caustic reaction mixture out of the flask as it boils. Gentlyheat the solution until it just boils (hotplate set at a maximum of 3 or 4 on the dial). If the watch glass rattles violently on top of the flask, then you have the hotplate set too high; turn it down. Continue gently heating for 20 minutes.
Remove the flask from the hotplate and allow it to cool to room temperature. Carefully add 3 M H2SO4 to the solution with stirring until the solution is acidic (test by touching the solution with your glass stirring rod, and then touching the glass rod to pH paper). As much as 40 mL of the sulfuric acid solution may be required to make the solution acidic. Once the pH paper indicates a slightly acid solution, add an extra 5 mL of the 3 M H2SO4. The white product should precipitate at this point. Cool the mixture in an ice bath to assure complete precipitation. Recover the product by vacuum filtration, using a Büchner funnel. Rinse the solid product well with ice cold water, in the Büchner funnel, with the suction flowing.
Save approximately 10 mg of the product in a small container (such as a vial or crucible) and cover with a Kimwipe to dry. Recrystallize the remainder of the product from water.3-5 (More Info) Add about 100 mL water (it is better to use less than 100 mL than more than 100 mL), magnetic stir bar or a boiling stone and your crude product to a 250 mL beaker. Heat the mixture until of the product dissolves. If the mixture boils and the solid has not dissolved, carefully add small portions of water, returning the solution to a boil between additions, until it does dissolve. Remove the solution from the heat using a paper towel as a hot pad (as your instructor will demonstrate). Place the beaker on a paper towel and cover it with your watch glass. Allow the solution to cool slowly without disturbance. Needle-like crystals should form fairly quickly (if they don’t try scratching the bottom of the beaker with your glass stirring rod). When the solution has returned to room temperature (cool to the touch), vacuum filter the solution to recover the crystals of compound X. Transfer the product from the Büchner funnel to a watch glass, cover with a paper towel and place it in your desk to dry. (More Info)
Before coming to laboratory prepare a table in your notebook to record your titration data. This table should include places to enter the buret readings, the volume and molarity of the titrant used, and the calculated values of the quantities sought (Help Me). Remember that you will be doing two sets of titrations (one to standardize the NaOH and one to determine the molar mass of compound X) and that each titration should have its own table.
During this laboratory period you will be doing three things: measure the melting point of the pure and impure compound X, titrate compound X to determine its molar mass and interpret compound X‘s NMR spectrum. Each of these tasks takes some time, so it is imperative that you plan what you are going to do in lab so that you use your time wisely.
The first thing to do once you are in the laboratory is determine the mass of your dry product and record it in the Results section of your notebook.
Determining the Melting Point of Compound X
Pack a melting point capillary with enough compound X so that the bottom 2-3 mm of the tube are filled. Also prepare a separate sample of impure compound X. Be sure you know which is which. Your instructor will demonstrate the correct method for filling a melting point capillary.
Check that the Mel-Temp is at room temperature before proceeding. Never place a sample into a hot Mel-Temp. Carefully, place the melting point capillary in the Mel-Temp’s stage. If it doesn’t go in easily, do not force it. Look through the magnifying lens on the front of the Mel-Temp to make sure that the capillary is seated properly. There are three sample slots on the stage, which means you can run up to three samples simultaneously (this requires extra vigilance). Click here for more information on Mel-Temp operation.
Switch on the apparatus and start heating the sample by turning the knob on the front of the Mel-Temp to “4”. CAUTION! The capillary and the sample stage will become very hot during the course of this experiment. The pure product’s melting point range is reported to be 158-160 °C, and at the recommended setting it will take about 10 min for the Mel-Temp to reach this temperature. During the first few minutes you do not need to monitor the sample closely, but you must not forget about it. When the temperature reaches about 130 °C, decrease the power level so that the temperature change is about 1 °C per minute. This heating rate generally gives the most accurate melting point ranges.
As you pass 150 °C observe the sample more closely and record any changes in the its appearance. The temperature at which you observe the first drop of liquid forming is thestart of your melting point range. Continue closely monitoring the sample until the last bit of solid melts. This is the end of the melting point range. Record the melting point range in your notebook (e. g., 152.0-155.3 °C). If your sample is pure, your melting point range will be less than 0.1 °C (the melting point is said to be “sharp”). If you miss the start or the end of the melting point range, you will need to start over.
When you are finished, set the knob to “0” and shut off the Mel-Temp. Remove your sample and discard it in the broken glass box. Leave the thermometer in the Mel-Temp.
Testing the Acidity of Compound X
In a small test tube dissolve a small amount (~10 mg) of compound X in a 15% by volume aqueous ethanol solution. Test the acidity of the resulting solution by transferring a small amount of the solution to litmus paper using a stirring rod. An acidic solution will cause blue litmus paper to turn red, while a basic solution will turn red litmus paper blue. Verify that compound X is an acid.
Titration of Compound X
Preparation and Standardization of the Base Solution
Place approximately 30 mL of 1 M NaOH in a clean 600-mL beaker and add 270 mL of distilled water. Swirl, then cover with a watch glass. This will become your standard base solution, but it is not critical at this point if we know the exact concentration, because we will standardize it. Calculate the approximate concentration of the NaOH solution. Use this value to estimate the equivalence point of a titration where this solution is used to titrate 0.350 g of the weak acid potassium hydrogen phthalate ( chemical formulaC8H5KO4, abbreviated KHP) via the reaction below:
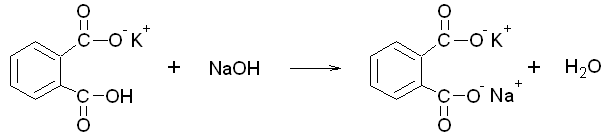
Scheme 4. The neutralization of potassium hydrogen phthalate (KHP) with NaOH.
Clean and rinse a buret with distilled water. Then rinse with three small portions (about 5 mL) of the base solution. Drain the rinse solution through the buret tip into a waste beaker between rinses. Fill the buret with the base solution and check for air bubbles in the tip. If there are any bubbles present, drain some solution through the tip by opening the buret’s stopcock. Refill the buret, if necessary and drain solution from the buret until the solution’s meniscus is below the 0.00 mL line on the buret. Record the initial buret reading to the nearest 0.01 mL.
Clean and rinse an Erlenmeyer flask (drying is not necessary). Mass and quantitatively transfer between 0.300 g and 0.400 g of KHP into the Erlenmeyer flask, and then add approximately 50 mL distilled water and 2 drops of phenolphthalein. Be sure to record the mass of the KHP to nearest milligram (by this we mean record the balance reading to the third decimal place).
Run approximately 50% of the volume that you calculated above to be the equivalence point from the buret into the flask as you swirl the flask. This can be done relatively rapidly, however if the solution turns pink and stays pink as you stir, stop the addition. Make a note of the approximate volume at which the color changed and start again, but this time use this volume as your target equivalence point.
As you approach your calculated equivalence point, add the NaOH solution from the buret at a slower rate as you continue to swirl the Erlenmeyer flask. As you near the end point of the titration the pink color will persist for a longer time. Slow the addition of the NaOH further as you near the calculated equivalence point. Add the NaOH solution dropwise with swirling until the first hint of a persistent (lasting more than 30 sec) pink color. If a drop is left hanging on the buret tip, gently touch the drop to the flask’s side to dislodge it and rinse it down into the solution with a little distilled water. Record the final volume of the NaOH solution in the buret to the nearest 0.01 mL.
Discard the titrated solution in the sink and then clean and rinse your flask (you don’t have to dry it!). Repeat this titration until you have three concentrations that agree within 2%. If you expect that enough NaOH solution remains in the buret to perform another titration, do not refill the buret. It saves time and it may increase the accuracy of your result. Note that this means that the final buret reading of one trial would be the initial buret reading of the next trial.
Titration of Compound X
Weigh accurately between 0.100 and 0.150 g of compound X, and place it in a clean 250-mL Erlenmeyer flask. Add approximately 50 mL of the 15% aqueous ethanol solution with swirling. Note that the solid will not dissolve completely, but that this will not matter. Add 2 drops of phenolphthalein indicator to the solution in the flask.
Refill the buret with the now standardized NaOH solution (the solution in the 600 mL beaker). Record the initial level of the NaOH in the buret. Titrate the solution of compound X. IMPORTANT! Compound X will dissolve as it reacts with the NaOH. You must be careful during the titration not to add too much NaOH at any one time and to constantly swirl the flask. From time to time rinse the sides of the flask with a little distilled water to dislodge any compound X crystals from the side. Ensure that all solid has dissolved prior to declaring the end point. From the volume of base used, calculate the molar mass of compound X (helpful hint: set these calculations up before lab). Repeat the titration (discarding the titrated solution and rinsing the Erlenmeyer between analyses) until you obtain three molar masses for compound X that agree with each other within 2%.
Results and Analysis
Melting Point Determination
Pure compounds have very sharp melting points (small melting point ranges). Based on this information, is your compound X pure?
Titration
Assume that compound X is a monoprotic acid (one H+ per molecule of X will react with NaOH), and calculate the molar mass of compound X from your titration results. Do this for each titration separately, then calculate an average molar mass and a standard deviation for the molar mass. Calculate the average molar mass for the class as a whole, perform a Q-test (if warranted), calculate the standard deviation of the class data and the confidence interval at the 95% confidence limit. Compare your result to that of the whole class.
NMR Spectral Analysis
Click here to view the 1H and 13C NMR spectra of compound X and click here to obtain the spectra in PDF format (suitable for printing). Using the information in Table 1 and the1H and 13C NMR spectra of methyl salicylate assign the spectra to the best of your ability. Here are some questions to ask as you examine the spectra: 1) how many peaks are there? 2) have you lost any relative to those you started with in methyl salicylate? 3) have you gained any peaks relative to methyl salicylate? 4) what are the similarities between compound X‘s NMR spectra and those of methyl salicylate? Look carefully at the integration in the 1H spectrum, it will help you.
Once you have assigned the NMR, you should be able to draw a structure (like that of methyl salicylate shown in Fig. 1) of compound X. You should also have a direct measure of the number of C and H atoms (and by default O atoms, too) in compound X. Calculate the molar mass of compound X based on this information and compare it to your titration results.
Since you know the molar mass and chemical formula of compound X, you can balance the chemical equation shown in Scheme 2 (hint: there is only one other product in addition to compound X, use the Law of Conservation of Mass to determine its formula). And with the balanced chemical equation you can calculate a percent yield for the reaction. Share your percent yield with others in the class.
Conclusions
This exercise is primarily a synthesis experiment; use the basic outline for a synthesis experiment as you write your conclusions. When you discuss the properties of the prepared material (section A of the synthesis experiment’s conclusions), give a brief outline of how you assigned the NMR spectra and used them to identify compound X. You are also to discuss your titration results (follow section A of the outline for a measurement exercise) and the melting point range. What must be made clear to the reader is how each of these means of characterization allowed you to identify compoundX; your logic is important!
- 1. Click here to download a copy of this document in PDF format. Note that hyperlinks are not active in the pdf version.
- 2. Ege, S. Organic Chemistry; Heath: Lexington, MA; 1984, p. 678 and p. 703.
- 3. Mohrig, J. R.; Hammond, C. N.; Morrill, T. C.; Neckers, D. C. Experimental Organic Chemistry; Freeman: New York; 1998, p. 32 36.
- 4. Pavia, D. L.; Lampman, G. M.; Kriz, G. S., Jr. Introduction to Organic Laboratory Techniques: A Contemporary Approach, 2nd Ed.; Saunders: New York; 1982, 93-94.
- 5. Byrd, H. and O’Donnell, S. E. J. Chem. Educ. 2003, 80, 174-176.